Proteins are vital molecules in the functioning of cells, carrying out various processes essential for life. To perform their functions accurately, proteins must have the correct three-dimensional structure. Recently, the HUN-REN-ELTE Protein Modeling Research Group introduced a groundbreaking method in Nature Communications called LoCoHD (Local Composition Hellinger Distance), which allows for the comparison of protein structures using not only spatial information but also chemical properties.
The LoCoHD Algorithm
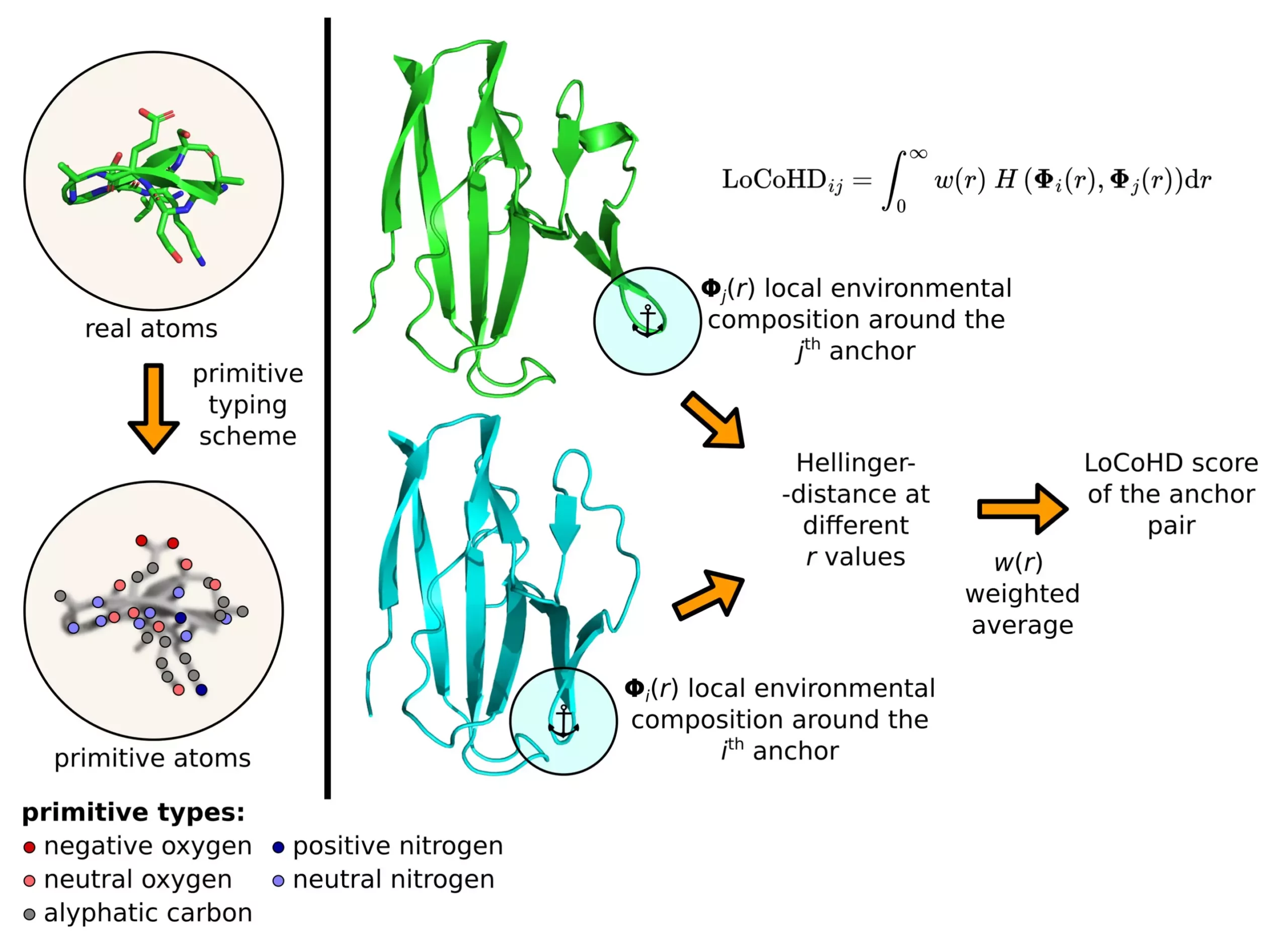
The LoCoHD algorithm, developed by Zsolt Fazekas, focuses on comparing the local environments around amino acids in proteins based on their chemical nature. It assigns a numerical value between 0 to 1 to indicate the similarity or dissimilarity of the compared structures. This metric provides valuable insights into the differences in atomic arrangements and chemical properties of proteins, aiding in understanding their functions better.
Methodology
The algorithm employs a multi-step protocol to generate the structural differences between proteins. Firstly, it converts real atoms into primitive atoms, representing their chemical nature. These primitive atoms act as reference points for comparison, with selected pairs called anchor atoms. The dissimilarity measure between these anchor atom pairs is computed to determine the overall difference between the structures. This method provides a comprehensive analysis of protein structures at a local and global level, facilitating detailed insights into their conformational changes.
The LoCoHD method has significant implications in the field of protein research, particularly in the Critical Assessment of Protein Structure Prediction (CASP) competitions. By incorporating chemical information into structural comparisons, the algorithm offers a more comprehensive evaluation of predicted protein structures. In the latest CASP14 competition, researchers utilized the LoCoHD method to compare modeled proteins, including structures predicted by advanced techniques like AlphaFold2.
Researchers employed the LoCoHD method to analyze proteins with significant biological relevance, such as the ORF8 protein from the SARS-CoV-2 virus. By identifying unique amino acid environments in protein models, the algorithm highlighted discrepancies in interaction patterns compared to experimental structures. Furthermore, the study extended to dynamic analyses of proteins, including the podocin protein involved in kidney function and the HIV-1 capsid protein critical for viral envelope formation. The method successfully identified key amino acids undergoing chemical changes during protein movement, aiding in understanding structural dynamics and function.
The insights provided by the LoCoHD method not only contribute to fundamental research in protein structures but also have practical implications for drug development. By understanding the chemical environments of proteins involved in disease pathways, researchers can target specific regions for therapeutic intervention. The detailed analysis facilitated by the algorithm enhances our understanding of disease mechanisms and helps in developing more effective treatments.
The LoCoHD method represents a significant advancement in protein structure comparison, bridging the gap between spatial conformation and chemical properties. By integrating these aspects, researchers can gain deeper insights into protein functions, enabling innovative approaches in both basic research and drug discovery.
Leave a Reply