The pharmaceutical industry heavily relies on the physical properties of pharmaceutical and functional materials to determine their performance. These properties, such as stability and solubility, are known to depend on factors like the solid-state form and environmental conditions. Late appearing, more stable forms can lead to the disappearance of polymorphs and potentially the withdrawal of life-saving medicines from the market. To mitigate these risks, the industry has invested in solid form screening platforms. However, quantitatively measuring the free energy differences between crystalline forms has proven to be a significant challenge.
Metastable crystal forms, although important for understanding the physical stability of materials, are often difficult to prepare in pure form and susceptible to converting into more stable forms. Consequently, accurately predicting free energies and understanding physical instability for all systems becomes crucial, especially for experimentally intractable cases. However, lack of reliable experimental data has hindered the development of computational methods for predicting solid-solid free energy differences. Most of the experimental data on free energy determinations for pharmaceutical molecules remains unpublished.
In a groundbreaking study published in the journal Nature, experts from academia and industry have compiled the first-ever reliable experimental benchmark of solid-solid free energy differences for chemically diverse, industrially relevant systems. This benchmark provides a valuable foundation for further research and development in the field.
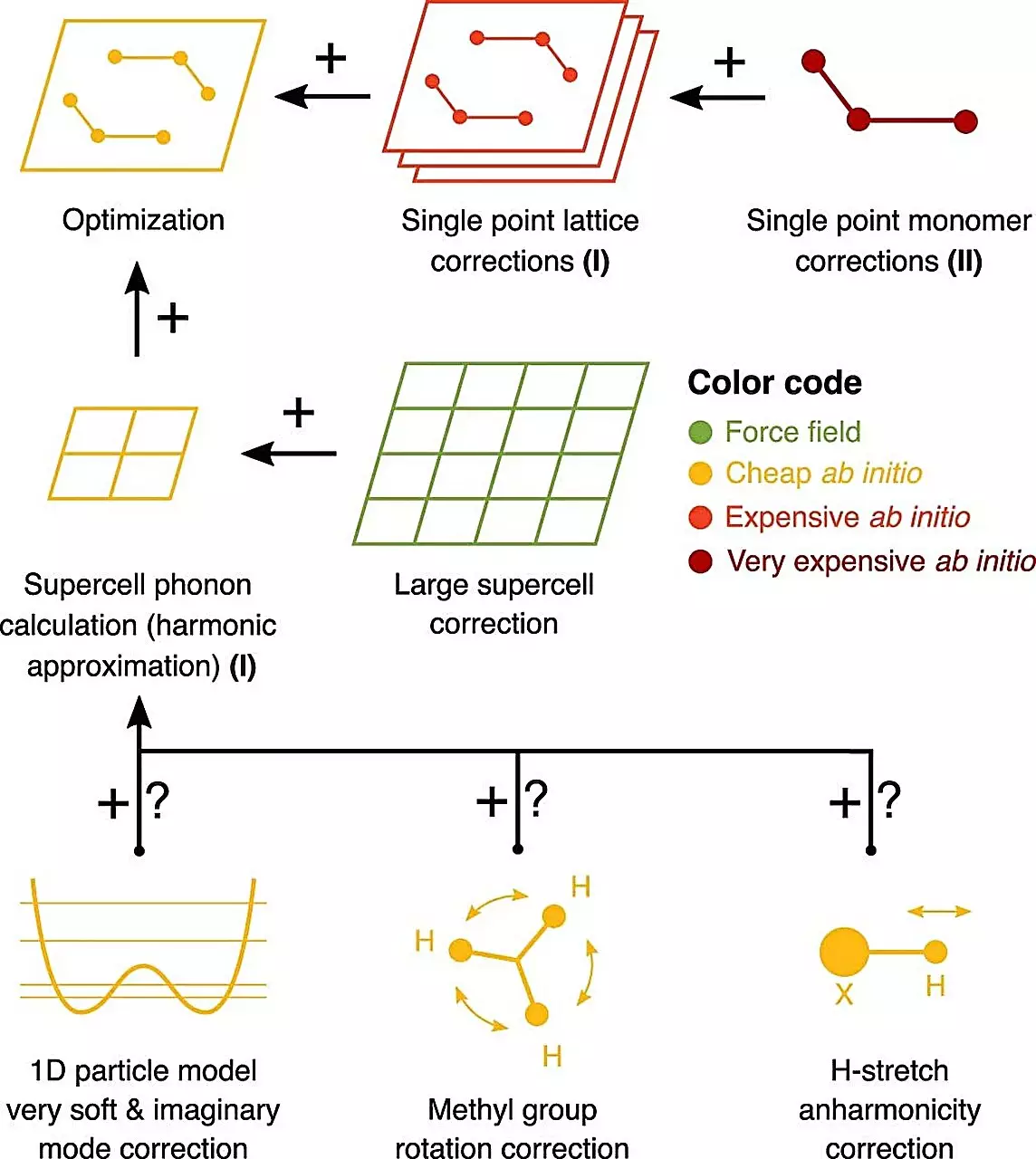
To overcome the challenge posed by the lack of experimental data, a group of researchers led by Prof. Alexandre Tkatchenko from the University of Luxembourg and Dr. Marcus Neumann from Avant-garde Materials Simulation (AMS) employed computational methods to predict these free energy differences. The calculations were performed using high-performance computing (HPC) and did not rely on any empirical input.
Remarkably, the computational predictions made by these methods showed surprising accuracy when compared to the experimental data from seven pharmaceutical companies. Without the need for empirical data, these computational models were able to predict and explain the energetics of drug crystal forms. This breakthrough demonstrates the potential of quantum mechanical calculations in revolutionizing the pharmaceutical industry.
The implications of this work are manifold. The ability to computationally model free energies opens up new avenues for understanding and mitigating risks associated with physical instability in pharmaceutical materials. By accurately predicting stable crystal forms and identifying potential polymorphic transformations, researchers can design more stable and effective drugs. This breakthrough also highlights the potential application of computational methods in various aspects of pharmaceutical research, from drug discovery to formulation optimization.
This significant achievement is the result of collaborative efforts between academia and industry. The methods developed by Prof. Tkatchenko’s academic group have been quickly adopted by the pharmaceutical industry, breaking the traditional barrier between research and industrial innovation. This collaboration has created an environment that promotes creativity and fosters core values such as honesty, integrity, perseverance, team spirit, and a genuine care for people and the environment.
Building bridges between fundamental science, high-performance computing, and major industry players is a remarkable accomplishment. The application of quantum mechanical calculations in pharmaceutical research has the potential to make a lasting impact on the future of healthcare. By leveraging computational methods, researchers can accelerate drug development, improve drug stability, and ensure the availability of life-saving medicines.
The development of computational methods to predict solid-solid free energy differences is revolutionizing pharmaceutical research. This breakthrough allows researchers to overcome the challenges posed by physical instability and polymorphism in pharmaceutical materials. By accurately predicting stable crystal forms, researchers can design more stable and effective drugs. The collaborative efforts between academia and industry have paved the way for innovative solutions that bridge the gap between research and industrial applications. With the potential to make a lasting impact on healthcare, computational methods are shaping the future of pharmaceutical research.
Leave a Reply