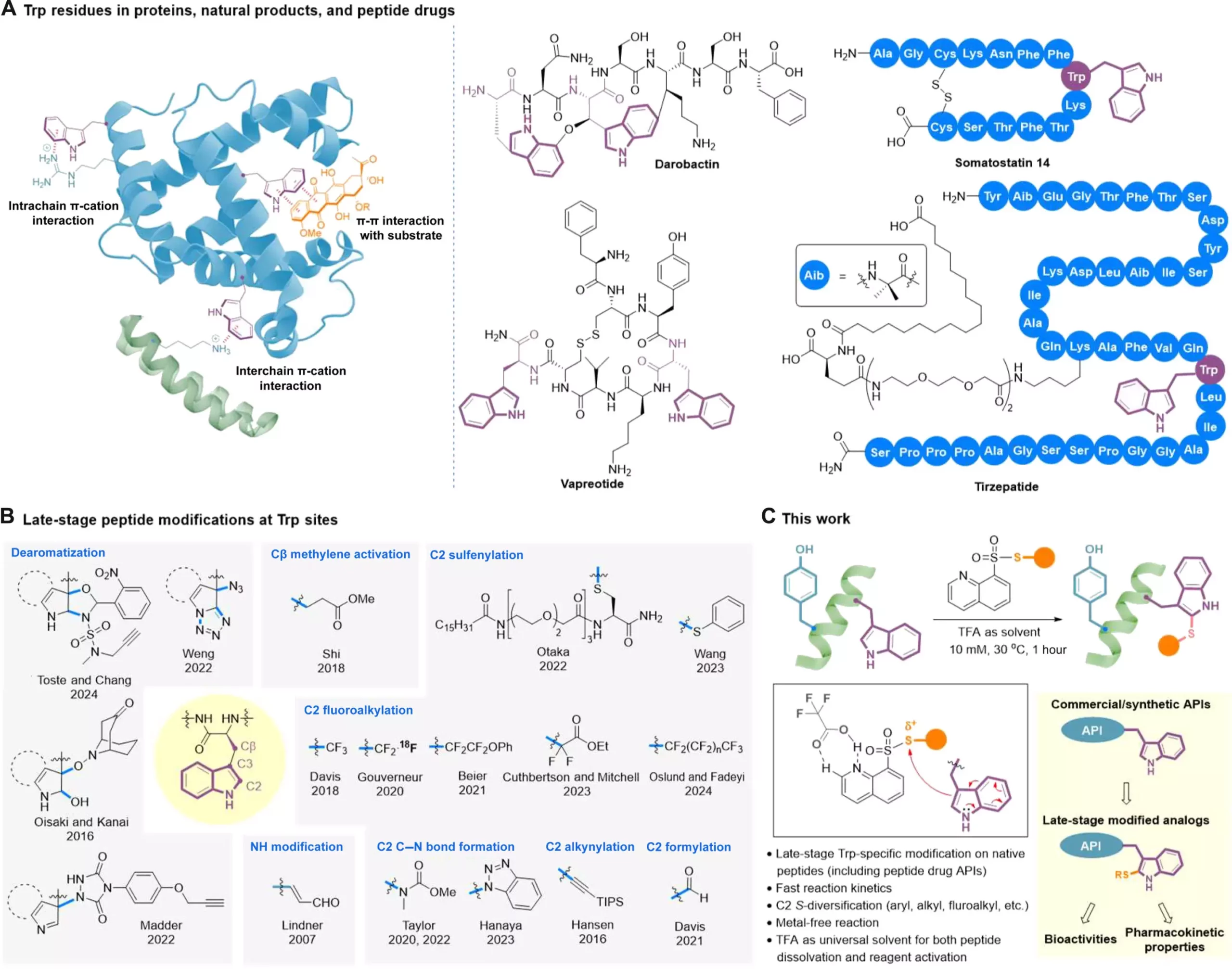
Peptides are gaining recognition as valuable therapeutic agents for tackling various medical challenges. Unlike small-molecule drugs, peptides have the advantage of targeting intricate biological processes with greater precision. They also tend to be less complex and more cost-effective compared to larger biological drugs like antibodies. Over 100 peptide drugs have received FDA approval, with around 40 of them containing tryptophan (Trp) residues, a crucial amino acid, in their molecules. The modification of these Trp residues has the potential to enhance drug-target interactions, as well as improve drug stability, bioavailability, and pharmacokinetics.
Despite the promising potential of peptide drugs, their dense functional nature presents challenges for achieving targeted modifications. Selectivity issues, such as chemoselectivity, regioselectivity, and stereoselectivity, must be addressed to enable effective transformations on peptide molecules. Additionally, the nucleophilic properties of peptides make them sensitive to redox conditions, further complicating the modification process. The limited solvents available for dissolving peptides without protection add another layer of difficulty, making site-specific late-stage modifications a daunting task.
A recent development by Professor Xuechen Li’s team at The University of Hong Kong introduces a clickable tryptophan modification strategy to address these challenges. This innovative approach allows for the modification of specific regions within a peptide molecule even in the later stages of drug development. By utilizing a catalyst-free C2-sulfenylation reaction with S-modified quinoline-containing thiosulfonate reagents, the researchers successfully incorporated various functional groups into Trp residues in native peptide structures. These groups include trifluoromethylthio, difluoromethylthio, (ethoxycarbonyl) difluoromethylthio, alkylthio, and arylthiol.
The use of trifluoroacetic acid (TFA) as a solvent proved crucial in activating the reagents through hydrogen bond interactions. TFA’s exceptional dissolving capabilities for hydrophobic and aggregation-prone peptides make this method applicable even to challenging molecules like lipopeptides and self-assembling peptides at higher concentrations. The team successfully applied this approach to modify several FDA-approved peptide drugs, showcasing its versatility in diversifying peptide-based active pharmaceutical ingredients. The method not only improved the bioactivity and serum stability of modified analogs but also demonstrated its potential in drug development.
With Trp being prevalent in various natural products and drug leads, this late-stage modification technique opens up possibilities for diversifying molecular libraries and functional probes. Medicinal chemists, peptide chemists, and chemical biologists stand to benefit from this single-step clickable method for generating structural analogs efficiently and cost-effectively. The platform created by Professor Li’s team represents a significant advancement in optimizing drug activities and pharmacokinetic properties, paving the way for innovative developments in the field of peptide drug development.
Leave a Reply