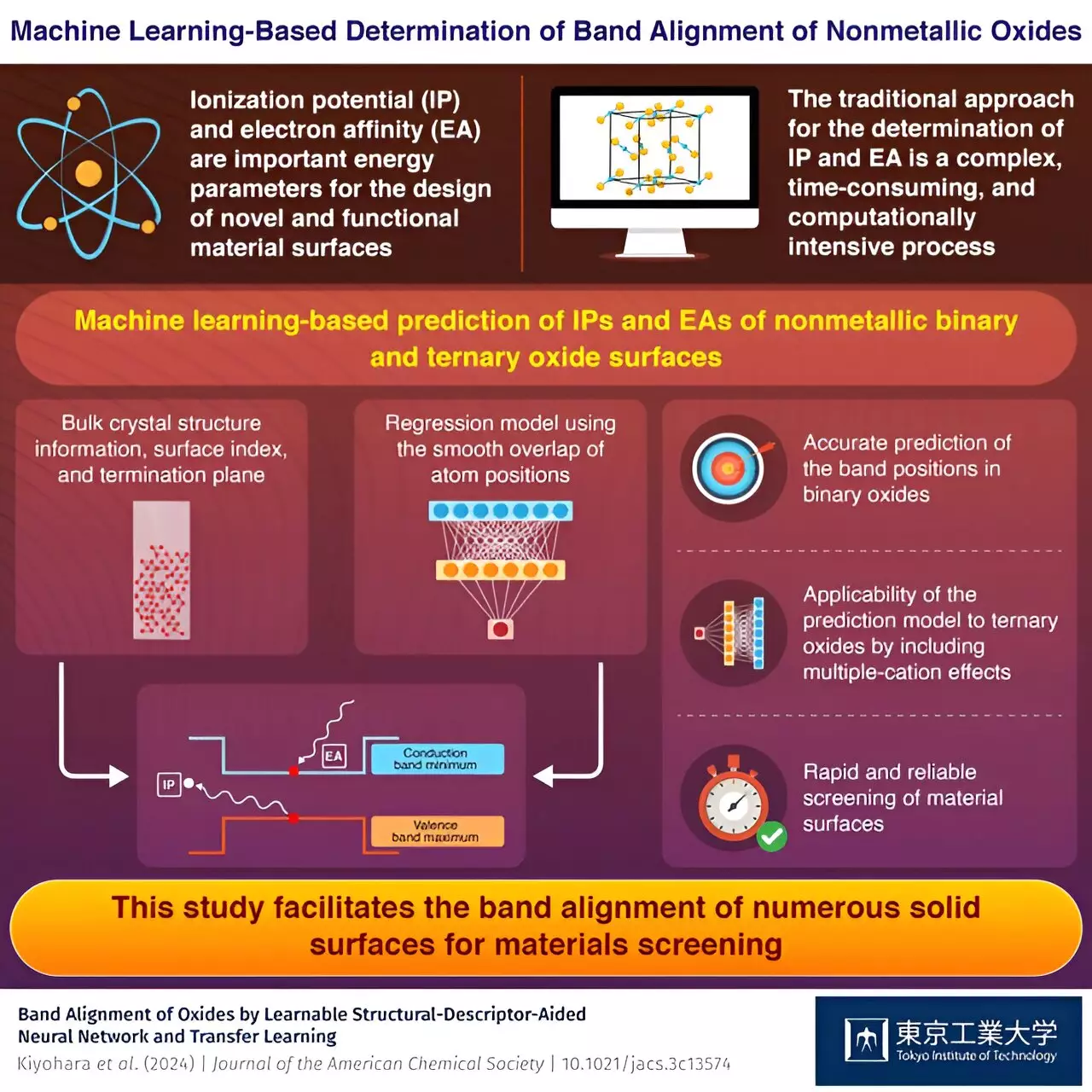
The field of materials science has seen significant advancements in recent years, with machine learning (ML) playing a crucial role in predicting electronic properties of various compounds. Scientists from Tokyo Tech have demonstrated the effectiveness of ML in accurately estimating fundamental electronic properties of binary and ternary oxide surfaces, paving the way for the development of novel materials with superior functionalities.
Understanding the atomic and electronic structures of materials is essential for designing and developing functional materials with desirable properties. Parameters such as ionization potential (IP) and electron affinity (EA) provide valuable insights into the electronic band structure of surfaces, making them crucial for applications in photosensitive equipment and optoelectronic devices. However, quantifying these properties accurately for nonmetallic materials poses challenges due to the complex nature of their surface structures.
Traditional approaches for computing IPs and EAs rely on labor-intensive first-principles calculations, which can be time-consuming and impractical for a wide range of surfaces. This limitation calls for the development of more efficient methods that can provide accurate predictions of electronic properties without sacrificing computational cost.
The team of scientists from Tokyo Tech, led by Professor Fumiyasu Oba, turned to ML as a solution to the challenges associated with traditional computation methods. ML offers the ability to train large datasets and predict electronic properties with high accuracy, making it an attractive tool for materials science research. By leveraging an artificial neural network and incorporating smooth overlap of atom positions (SOAPs) as input data, the researchers were able to develop a regression model that accurately predicted IPs and EAs of binary oxide surfaces.
In addition to binary oxides, the ML-based model demonstrated the capability of predicting electronic properties of ternary oxides by incorporating information on bulk crystal structures and surface termination planes. The concept of “transfer learning” further enhanced the model’s predictive capabilities, enabling it to adapt to new datasets and tasks seamlessly. By developing “learnable” SOAPs and considering the effects of multiple cations, the researchers successfully predicted the IPs and EAs of ternary oxides, showcasing the versatility and efficiency of ML in materials science research.
The integration of machine learning in predicting electronic properties of materials represents a significant advancement in materials science research. The work done by the team of scientists from Tokyo Tech highlights the potential of ML in accelerating the discovery and development of novel materials with superior functionalities. By combining theoretical calculations with ML algorithms, researchers can overcome the challenges associated with traditional computation methods and explore new frontiers in materials design and engineering.
Leave a Reply